| Sommaire |
|---|
Trimmomatic
Trimmomatic [1] est un outil pour "trimmer" les données FASTQ d'Illumina et enlever les séquences de faibles qualités, ainsi que celle de l'adaptateur. Au cours du séquencage avec Illumina, des séquences adaptatrices sont ajoutées de part et d'autres de chaque fragment. Il est important de retirer ces séquences avant toute analyse ultérieure, puisqu'elles risquent de fausser l'assemblage et les résultats des analyses. Par ailleurs, la méthode de séquencage par synthèse employée par Illumina peut résulter en une accumulation d'erreurs aux extrémités des reads [2]. On assiste donc à une variation de la qualité des séquences le long des reads. Grâce à Trimmomatic, les régions de faible qualités peuvent être trimmées, de même que les séquences contaminantes. Trimmomatic est donc l'une des premières étapes pour contrôler la qualité de vos reads.
Le programme est installé sur les serveurs du M5. Pour accéder à l'aide, utiliser :
trimmomatic --help
PE [-threads <threads>] [-phred33|-phred64] [-trimlog <trimLogFile>] [-quiet] <input read 1> <input read 2> <paired output 1> <unpaired output 1> <paired output 2> <unpaired output 2> ILLUMINACLIP:<Adapter>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold> [OPTIONS DE TRIMMING]
SE [-version] [-threads <threads>] [-phred33|-phred64] [-trimlog <trimLogFile>] [-quiet] <inputFile> <outputFile> ILLUMINACLIP:<Adapter>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold> [OPTIONS DE TRIMMING]Trimmomatic propose deux modes : un mode pour les reads pairés (PE) et un autre pour les reads non-pairés (SE). Vous trouverez ci-dessous la liste des options utiles de Trimmomatic.
-threads NTHREADS: nombre de threads à utiliser-phred33 | -phred64: type de score de qualité à utiliser. Il est important de choisir le bon type d'encodage des scores-trimlog LOGFILEenregistrer trace dans un fichier.ILLUMINACLIP:<Adapter>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>Utilisation d'adaptateurs de type Illumina. Ces paramètres sont ceux utilisés pour la détection de la séquence de l'adaptateur. Trimmomatic détecte ces séquences en considérant un seuil (défini par l'utilisateur) pour leur score d'alignement local avec les reads puis en coupant la séquence du reads dans le sens 3'. Il est donc important que les seuils soient assez stringent pour éviter les faux positifs.
<Adapter>: Fichier fasta contenant l'adaptateur<seed mismatches>: Nombre maximum de mismatch pour considérer un alignement valide<palindrome clip threshold>: Précision du match entre deux adaptateurs pour le mode palindrome. Ce mode n'est valide que pour les reads pairés et permet d'identifier la contamination de séquence causée par une extension des reads avec la séquence de l'adaptateur (adapter read-through)<simple clip threshold>: Précision du match entre adaptateur et la séquence du read
OPTIONS DE TRIMMING: L'ordre des options que vous employez compte.LEADING:<seuil de qualité>: Couper les bases au début de la séquence des reads si leur qualité est inférieure au seuil définiTRAILING:<seuil de qualité>: Couper les bases à la fin de la séquence des reads si leur qualité est inférieure au seuil définiCROP:<longueur>: Couper les reads en enlevant les bases à la fin pour qu'ils aient la longueur spécifiée, peut importe la qualité des bases.HEADCROP:<longueur>: Couper les reads en enlevant le nombre spécifié de base au début de la séquenceSLIDINGWINDOW:<Taille de la fenêtre>:<seuil de qualité>: Utiliser une approche de fenêtre coulissante de l'extrémité 5' au 3' pour trimmer les reads. Trimmer dès que la qualité moyenne de la fenêtre descend en dessous d'un seuil défini.MINLEN:<longueur>: Enlever tous les reads dont la taille n'atteint pas la longueur requiseAVGQUA:<seuil de qualité>: Enlever le read si sa qualité moyenne est inférieur au seuil défini
Rcorrector
Rcorrector (RNA-seq error CORRECTOR) [3] est un outil de correction des erreurs aléatoires de séquencage au sein des données RNA-seq d'Illumina, qui utilise une approche basée sur les kmers. En effet, les erreurs de séquencage au sein des reads déja très courts, influencent la justesse des analyses ultérieurs effectuées sur ces reads, en particulier l'alignement et l'assemblage). Contrairement aux méthodes de correction des reads WGS (Whole Genome Sequencing) , la correction des reads de données RNA-seq fait face à plusieurs complications, en raison de la variation du niveau d'expression des gènes et de la présence de mécanismes tels que l'épissage alternatif, ce qui heureusement n'est pas le cas dans notre expérience.
Rcorrector utilise un graphe de De Bruijn pour représenter tous les k-mers présents au sein des reads et calcule un seuil local à chaque position du reads, pour chaque k-mer afin de le considérer comme valide ou non. En fonction du seuil défini et du chemin minimisant le nombre d'erreur, Rcorrector corrige les reads de façon adéquate.

Les k-mers sont déterminés avec JellyFish, un package très rapide, disposant d'une excellente optimisation de la mémoire pour les occurences des k-mers au sein des fragments d'adn.
Rcorrector est disponible sur les serveurs du M5. Pour accéder à l'aide, faites :
run_rcorrector.pl ou rcorrector
run_rcorrector.pl -s <seqfiles> -k <taille du kmer> -t <nombre de thread> -maxcorK <correction maximal> [-od <dossier output>]-s <seqfiles>: fichiers contenant les séquences, séparés par une virgule. Il est aussi possible d'utiliser-s file1 -s file2 ...-k <longueur du kmer>: taille du kmer à utiliser, doit être inférieur ou égale à 32. Pour le type de données dont nous disposons, il est recommandé d'utiliser 32.-t <nombre de threads>: nombre de threads à utiliser, il n'est pas recommandé d'utiliser plus de threads quil y a de processeurs-maxcorK <maximum correction>: nombre maximal de correction pour chaque read. 1 seule correction maximale par read est recommandée.-od <dossier outputr>: Chemin vers le dossier output. Par défaut les fichiers sont enregistrés dans le dossier courant (./).
Rcorrector retourne les reads corrigés dans des fichiers *.cor.fq.
L'entête des fichiers corrigés comprend l'information additionnelle suivante :
- "cor": indiquant que la séquence a été corrigée
- "unfixable_error": qui signifie que Rcorrector n'a pas pu corrriger les erreurs au sein de la séquence du read.
- "l: m: h:" désignant respectivement le plus petit, le median et le plus grand nombre de kmer au sein de la séquence du read correspondant.
Lighter
Lighter [4] est un autre outil de correction d'erreurs de séquençage WGS également basé sur les kmers. Toutefois, Lighter utilise l'échantillonage des kmers et non leurs occurences pour corriger les reads. Lighter présente l'avantage d'être extrêmement rapide

Pour utiliser lighter, ajouter la ligne suivante à la fin de votre .bashrc
setenv PATH ${PATH}:/usagers_bac/dbcm2002/espace/dbcm2002/PROG
Vous pouvez maintenant faire : lighter
lighter -r <seqfiles> -K <taille du kmer> <taille_du_génome> -t <nombre de thread> -maxcor <correction maximal> [-od <dossier output>]-r <seqfiles>: fichiers contenant les séquences. Il est possible d'utiliser-r file1 -r file2 ...-k <longueur du kmer> <taille_du_génome>: taille du kmer à utiliser (doit être inférieur ou égale à 32) et taille du génome. Pour le type de données dont nous disposons, il est recommandé d'utiliser 32 pour la longeur des kmers et 5M pour la taille du génome.-t <nombre de threads>: nombre de threads à utiliser, il n'est pas recommandé d'utiliser plus de threads qu'il y a de processeurs-maxcor <maximum correction>: nombre maximal de correction pour chaque read. 1 seule correction maximale par read est recommandée.-od <dossier outputr>: Chemin vers le dossier output. Par défaut les fichiers sont enregistrés dans le dossier courant (./).
Lighter retourne les reads corrigés dans des fichiers *.cor.fq.
L'entête des fichiers corrigés est similaire à celle de Rcorrector et comprend l'information additionnelle suivante :
- "cor": indiquant que la séquence a été corrigée
- "unfixable_error": qui signifie que les erreurs au sein de la séquence du readsn'ont pas pu être corrigé.
- "bad_suffix", "bad_prefix": indique que l'erreur dans le suffixe ou le préfixe n'a pas pû être corrigée.
SPAdes
SPAdes (St. Petersburg genome assembler) [4] est un assembleur de génome bactérien, écrit en python, qui fonctionne avec les données Illumina, IonTorrent et PacBio. Outre l'assemblage, le pipeline de SPAdes propose également plusieurs autres modules dont la correction de reads.
SPAdes utilise un algorithme original basé sur les graphes de de Bruijn pairés (PDBG) [5] avec plusieurs ajustements pour rendre l'algorithme plus praticable en présence d'erreurs au sein des reads.

SPAdes prends comme input, les reads pairés ou non-pairés (Illumina, 454, PacBio) en format fasta ou fastq. Le programme est installé sur les serveurs du M5.
Il faut tout d'abord charger le module avant toute tentative d'exécution.
module load spades
Pour accéder à l'aide, utilisez ensuite :
spades.py --help.
Une ligne de commande typique d'exécution de SPAdes ressemble donc à ceci :
spades.py [input] [option du pipeline] [options avancées] -o outdir [input] : Fichier fastq des reads pairés ou non-pairés.
...
[option du pipeline] : Il s'agit des options reliées à l'exécution du pipeline.
--only-error-correction: n'exécuter que le module de correction d'erreurs. L'assemblage n'est donc pas effectué.--only-assembler: ne faire que l'assemblage. Le module de correction n'est donc pas exécuté.--careful: réduit au maximum le nombre de mismatches et d'indel courts. Cette option demande un temps d'exécution plus long.--continue: permet de continuer à partir du checkpoint le plus récent lorsque l'éxécution précédente a été coupée.
...
[options avancées] : utilisation de paramètres plus avancés.
-t <threads>: spécifiez le nombre de threads à utiliser--cov-cutoff <float>: seuil minimum requis pour la couverture.-k <int, int, int>: liste d'entiers impairs, en ordre croissant, correspondant à la taille des kmers. La taille maximale est fixée à 127.-m <int>: limite de la mémoire à utiliser par SPAdes en gb (250 par défaut). Si cette value est fixée, SPAdes arrêtera l'exécution dès que la mémoire utilisée dépasse la limite fixée.
...
-o <répertoire de sortie> : répertoire de sauvegarde des outputs de SPAdes.
Contrôle qualité des données Illumina
FastQC
FastQC est un programme JAVA de control de qualité de données de séquençage. FastQC utilise plusieurs métriques pour évaluer la qualité des séquences. Un rapport (html) est produit à la fin de l'analyse. Ce rapport très simple permet d'identifier les types d'erreurs potentiels au sein des données, donnant ainsi une idée sur leur origine. Le programme peut être exécuté soit en ligne de commande, soit à partir de l'interface graphique.
Pour accéder à l'aide, utiliser :
fastqc --help
fastqc [-o répertoire de sortie] [-f fastq|bam|sam] [-t nombre de threads] [-c fichier de contaminant] sequence1 sequence2 ... sequenceN-o <repertoire de sortie>: spécifier le répertoire de sortie qui contiendra l'output du programme.-f <fastq | bam | sam>: spécifier le format des séquences en entrée-t <nombre de threads>: spécifier le nombre de threads à utiliser au cours de l'éxécution-c <fichier de contaminant>: spécifier un fichier fasta contenant les séquences contaminantes potentielles (y compris la séquence de l'adaptateur)
Nettoyage des données Illumina
Au cours de la préparation des librairies de séquençage Illumina, des séquences adaptatrices sont ajoutées de part et d'autres de chaque fragment. Ces séquences sont toujours présente lors du séquençage et peuvent donc être séquencées et se retrouver dans les jeux de données Illumina. Il est donc important de retirer ces séquences avant toute analyse ultérieure, puisqu'elles risquent de fausser l'assemblage et les résultats des analyses.
De plus, la méthode de séquençage par synthèse employée par Illumina peut induire certains artéfacts, donc une accumulation d'erreurs aux extrémités des reads [2]. Il est donc nécessaire d'enlever certaines régions aux extrémités des lectures afin de limiter la présence d'erreur dans les séquences puisqu'elles peuvent affecter négativement les analyser en aval. Cette étape est donc l'une des premières étapes pour contrôler la qualité de vos reads.
Fastp
fastp est un outil qui permet d'effectuer à la fois le nettoyage des données Illumina et d'effectuer le contrôle qualité avant et après le nettoyage. Il s'agit d'un outil très rapide capable de détecter automatiquement les adapteurs ainsi que de corriger les artéfacts typique de la technologie de séquençage employée.
Pour accéder à l'aide, utiliser :
fastp --helpLa commande minimale pour lancer fastp est :
fastp -i in.R1.fq.gz -I in.R2.fq.gz -o out.R1.fq.gz -O out.R2.fq.gzCependant, comme vous pouvez voir, fastp peut aussi être configuré de manière extensive. Vous trouverez donc ci-dessous les principales options de fastp.
Entrée/sortie :
--in1 <in1.fastq>: fichier contenant les lectures forward.--in2 <in2.fastq>: fichier contenant les lectures reverse.--out1 <out1.fastq>: fichier de sortie des lectures forward nettoyées.--out2 <out2.fastq>: fichier de sortie des lectures reverse nettoyées.--unpaired1 <unpaired.fastq>: fichiers contenant les lectures non pairées (contient forward et reverse)
Adapteurs :
--adapter_sequence <sequence>: séquence des adapteurs forward.--adapter_sequence_r2 <sequence>: séquence des adapteurs reverse
Nettoyage des lectures de faible qualité
--cut_front: activer le nettoyage des lectures de faible qualité--cut_window_size <window>: taille de la fenêtre coulissante (sliding window)--cut_mean_quality <quality>: qualité minimale requise pour conserver la séquence--length_required <length>: longueur minimale pour conserver une lecture
Trimmomatic
Trimmomatic est un autre outil pour nettoyer les données FASTQ produite par Illumina. Il permet notamment d'enlever les séquences des adapteurs et de faibles qualités. Le programme est installé sur les serveurs SENS.
Pour accéder à l'aide, utiliser :
trimmomatic --help
PE [-threads <threads>] [-phred33|-phred64] [-trimlog <trimLogFile>] [-quiet] <input read 1> <input read 2> <paired output 1> <unpaired output 1> <paired output 2> <unpaired output 2> ILLUMINACLIP:<Adapter>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold> [OPTIONS DE TRIMMING]
SE [-version] [-threads <threads>] [-phred33|-phred64] [-trimlog <trimLogFile>] [-quiet] <inputFile> <outputFile> ILLUMINACLIP:<Adapter>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold> [OPTIONS DE TRIMMING]Trimmomatic propose deux modes : un mode pour les reads pairés (PE) et un autre pour les reads non-pairés (SE). Vous trouverez ci-dessous la liste des options utiles de Trimmomatic.
-threads NTHREADS: nombre de threads à utiliser-phred33 | -phred64: type de score de qualité à utiliser. Il est important de choisir le bon type d'encodage des scores-trimlog LOGFILEenregistrer trace dans un fichier.ILLUMINACLIP:<Adapter>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>Utilisation d'adaptateurs de type Illumina. Ces paramètres sont ceux utilisés pour la détection de la séquence de l'adaptateur. Trimmomatic détecte ces séquences en considérant un seuil (défini par l'utilisateur) pour leur score d'alignement local avec les reads puis en coupant la séquence du reads dans le sens 3'. Il est donc important que les seuils soient assez stringent pour éviter les faux positifs.<Adapter>: Fichier fasta contenant l'adaptateur<seed mismatches>: Nombre maximum de mismatch pour considérer un alignement valide<palindrome clip threshold>: Précision du match entre deux adaptateurs pour le mode palindrome. Ce mode n'est valide que pour les reads pairés et permet d'identifier la contamination de séquence causée par une extension des reads avec la séquence de l'adaptateur (adapter read-through)<simple clip threshold>: Précision du match entre adaptateur et la séquence du read
OPTIONS DE TRIMMING: L'ordre des options que vous employez compte.LEADING:<seuil de qualité>: Couper les bases au début de la séquence des reads si leur qualité est inférieure au seuil définiTRAILING:<seuil de qualité>: Couper les bases à la fin de la séquence des reads si leur qualité est inférieure au seuil définiCROP:<longueur>: Couper les reads en enlevant les bases à la fin pour qu'ils aient la longueur spécifiée, peut importe la qualité des bases.HEADCROP:<longueur>: Couper les reads en enlevant le nombre spécifié de base au début de la séquenceSLIDINGWINDOW:<Taille de la fenêtre>:<seuil de qualité>: Utiliser une approche de fenêtre coulissante de l'extrémité 5' au 3' pour trimmer les reads. Trimmer dès que la qualité moyenne de la fenêtre descend en dessous d'un seuil défini.MINLEN:<longueur>: Enlever tous les reads dont la taille n'atteint pas la longueur requiseAVGQUA:<seuil de qualité>: Enlever le read si sa qualité moyenne est inférieur au seuil défini
Rcorrector
Rcorrector (RNA-seq error CORRECTOR) [1] est un outil de correction des erreurs aléatoires de séquençage au sein des données RNA-seq d'Illumina, qui utilise une approche basée sur les k-mers. En effet, les erreurs de séquençage au sein des reads déja très courts, influencent la justesse des analyses ultérieurs effectuées sur ces reads, en particulier l'alignement et l'assemblage). Contrairement aux méthodes de correction des reads WGS (Whole Genome Sequencing) , la correction des reads de données RNA-seq fait face à plusieurs complications, en raison de la variation du niveau d'expression des gènes et de la présence de mécanismes tels que l'épissage alternatif, ce qui heureusement n'est pas le cas dans notre expérience.
Rcorrector utilise un graphe de De Bruijn pour représenter tous les k-mers présents au sein des reads et calcule un seuil local à chaque position du reads, pour chaque k-mer afin de le considérer comme valide ou non. En fonction du seuil défini et du chemin minimisant le nombre d'erreur, Rcorrector corrige les reads de façon adéquate.
Les k-mers sont déterminés avec JellyFish, un package très rapide, disposant d'une excellente optimisation de la mémoire pour les occurences des k-mers au sein des fragments d'ADN.
Rcorrector est disponible sur les serveurs du M5. Pour accéder à l'aide, faites :
Les principales options de Rcorrector sont décrites ci-dessous.
run_rcorrector.pl -s <seqfiles> -k <taille du kmer> -t <nombre de thread> -maxcorK <correction maximal> [-od <dossier output>]-s <seqfiles>: fichiers contenant les séquences, séparés par une virgule. Il est aussi possible d'utiliser-s file1 -s file2 ...-k <longueur du kmer>: taille du kmer à utiliser, doit être inférieur ou égale à 32. Pour le type de données dont nous disposons, il est recommandé d'utiliser 32.-t <nombre de threads>: nombre de threads à utiliser, il n'est pas recommandé d'utiliser plus de threads quil y a de processeurs-maxcorK <maximum correction>: nombre maximal de correction pour chaque read. 1 seule correction maximale par read est recommandée.-od <dossier outputr>: Chemin vers le dossier output. Par défaut les fichiers sont enregistrés dans le dossier courant (./).
Rcorrector retourne les reads corrigés dans des fichiers *.cor.fq ou *.cor.fq.gz.
L'entête des fichiers corrigés comprend l'information additionnelle suivante :
- "cor": indiquant que la séquence a été corrigée
- "unfixable_error": qui signifie que Rcorrector n'a pas pu corrriger les erreurs au sein de la séquence du read.
- "l: m: h:" désignant respectivement le plus petit, le median et le plus grand nombre de kmer au sein de la séquence du read correspondant.
Lighter
Lighter [2] est un autre outil de correction d'erreurs de séquençage WGS également basé sur les kmers. Toutefois, Lighter utilise l'échantillonage des kmers et non leurs occurences pour corriger les reads. Lighter présente l'avantage d'être extrêmement rapide
Pour utiliser lighter, ajouter la ligne suivante à la fin de votre .bashrc
setenv PATH ${PATH}:/usagers_bac/dbcm2002/espace/dbcm2002/PROG
Vous pouvez maintenant faire : lighter
lighter -r <seqfiles> -K <taille du kmer> <taille_du_génome> -t <nombre de thread> -maxcor <correction maximal> [-od <dossier output>]-r <seqfiles>: fichiers contenant les séquences. Il est possible d'utiliser-r file1 -r file2 ...-k <longueur du kmer> <taille_du_génome>: taille du kmer à utiliser (doit être inférieur ou égale à 32) et taille du génome. Pour le type de données dont nous disposons, il est recommandé d'utiliser 32 pour la longeur des kmers et 5M pour la taille du génome.-t <nombre de threads>: nombre de threads à utiliser, il n'est pas recommandé d'utiliser plus de threads qu'il y a de processeurs-maxcor <maximum correction>: nombre maximal de correction pour chaque read. 1 seule correction maximale par read est recommandée.-od <dossier outputr>: Chemin vers le dossier output. Par défaut les fichiers sont enregistrés dans le dossier courant (./).
Lighter retourne les reads corrigés dans des fichiers *.cor.fq.
L'entête des fichiers corrigés est similaire à celle de Rcorrector et comprend l'information additionnelle suivante :
- "cor": indiquant que la séquence a été corrigée
- "unfixable_error": qui signifie que les erreurs au sein de la séquence du readsn'ont pas pu être corrigé.
- "bad_suffix", "bad_prefix": indique que l'erreur dans le suffixe ou le préfixe n'a pas pû être corrigée.
SPAdes
SPAdes (St. Petersburg genome assembler) [3] est un assembleur de génome bactérien, écrit en python, qui fonctionne avec les données Illumina, IonTorrent, Oxford Nanopore et PacBio. Outre l'assemblage, le pipeline de SPAdes propose également plusieurs autres modules dont la correction de reads. SPAdes utilise un algorithme original basé sur les graphes de de Bruijn pairés (PDBG) [4] avec plusieurs ajustements pour rendre l'algorithme plus praticable en présence d'erreurs au sein des reads. La résolution de la séquence circulaire CATCAGATAGGA par cet algorithme pourrait ressembler à la figure suivante.

SPAdes prends comme input, les reads pairés ou non-pairés (Illumina, 454, PacBio, Oxford Nanopore) en format fasta ou fastq. Le programme est installé sur les serveurs SENS.
Pour utilise ce programme, il faut d'abord charger le module :
module load spades-3.15.4
Pour accéder à l'aide, utilisez ensuite :
spades.py --help.
Une ligne de commande typique d'exécution de SPAdes ressemble donc à ceci :
spades.py [input] [option du pipeline] [options avancées] -o outdir [input]: Fichier fastq des reads pairés ou non-pairés.--pe#-1 <reads pairés forward>: spécifie le fichier fastq contenant les reads forward pour les librairies paired-end. Le#est un chiffre entre [1-9] qui désigne le numéro de la librairie--pe#-2 <reads pairés reverse>: spécifie le fichier fastq contenant les reads reverse pour les librairies paired-end. Utilisez le même numéro de librairie lorsqu'il s'agit de données de la même expérience.--s# <reads non-pairés>: spécifie le fichier fastq contenant les reads non-pairés.
[option du pipeline]: Il s'agit des options reliées à l'exécution du pipeline.--only-error-correction: n'exécuter que le module de correction d'erreurs. L'assemblage n'est donc pas effectué.--only-assembler: ne faire que l'assemblage. Le module de correction n'est donc pas exécuté. Cette option est généralement utilisée si les lectures sont corrigées avec un autre outil (ex. Rcorrector ou Lighter).--careful: réduit au maximum le nombre de mismatches et d'indel courts. Cette option demande un temps d'exécution plus long.--continue: permet de continuer à partir du checkpoint le plus récent lorsque l'éxécution précédente a été coupée.
[options avancées]: utilisation de paramètres plus avancés.-t <threads>: spécifiez le nombre de threads à utiliser--cov-cutoff <float>: seuil minimum requis pour la couverture.-k <int, int, int>: liste d'entiers impairs, en ordre croissant, correspondant à la taille des kmers. La taille maximale est fixée à 127.-m <int>: limite de la mémoire à utiliser par SPAdes en gb (250 par défaut). Si cette value est fixée, SPAdes arrêtera l'exécution dès que la mémoire utilisée dépasse la limite fixée.
-o <répertoire de sortie>: répertoire de sauvegarde des outputs de SPAdes.
Les outputs de SPAdes se trouvent tous dans le dossier spécifié. La liste non-exhaustive suivante représente quelques uns des fichiers outputs de SPAdes :
before_rr.fasta: contient les contigs avant résolution du graphe d'assemblage.contigs.fasta: contient les contigs.scaffolds.fasta: contient les scaffolds.assembly_graph.fastg: contient le graphe d'assemblage de SPAdes en format FASTG. Il est possible de visualiser ce graph avec Bandage.contigs.paths: contient les chemins présents dans le graphe d'assemblage des contigs.scaffolds.paths: contient les chemins présents dans le graphe d'assemblage des scaffoldsparams.txt: information sur les paramètres de SPAdes utilisés au cours de l'éxécutionspades.log: SPAdes logdataset.info: fichier de configutation interneK<##>/: dossier contenant les fichiers correspondant à l'éxécution avec comme longueur de kmer <##>
Explication de l'entête des fichiers contigs.fasta et scaffolds.fasta
Les entêtes des fichiers fasta en output sont de la forme suivante : >NODE_7_length_217965_cov_40.4652_ID_4150. Le 7 désigne le numéro du noeud au sein du graphe (correspond à un contig ou un scaffold). 217965 désigne la longueur de la séquence, 40.4652 la couverture des kmers et 4150 l'ID unique associé au noeud.
Prokka
Prokka [6] est un pipeline écrit en perl pour l'annotation rapide de génomes prokaryotes. L'annotation des génomes permet l'extraction d'informations génétiques pertinentes de la séquence nucléotidique brute des génomes.
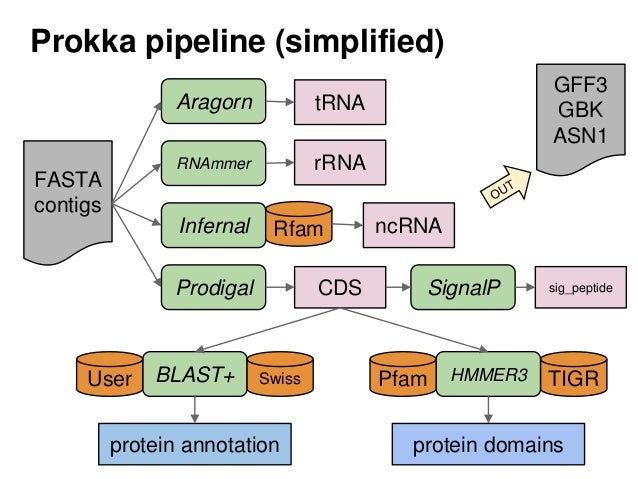
Prokka est installé sur les serveurs du M5. Vous devez tout d'abord charger le module avant toute tentative d'exécution.
module load prokka
La simplicité des commandes de Prokka permet de l'exécuter avec un mimimum de paramètres. Ainsi un simple ``prokka contigs.fa``` suffira pour annoter un génome d'intérêt à partir des contigs contenus dans le fichier contigs.fa. Il est toutefois possible de spécifier soi-même les paramètres, ce qui présente un avantage particulier pour les utilisateurs avancés. Prokka fourni également des modules pour la soumission des données sur Genbank et ENA.
Pour accéder à l'aide, il suffit de faire :prokka --help
prokka [options] <contigs.fasta>Vous trouverez ci-dessous une liste d'options fréquemment utilisées:
--kingdom <Archaea | Bacteria | Mitochondria | Viruses>: permet de spécifier le domaine à utiliser par Prokka parmi ceux proposés. Par défaut, Prokka assume qu'il s'agit d'un génome bactérien.--usegenus: permet d'indiquer s'il faut utiliser ou non une base de données BLAST spécifique à un genre taxonomic. Nécessite l'utilisation du paramètre--genus--genus <genre>: spécifier le genre taxonomic du génome s'il est connu.--species <espèce>: spécifier l'espèce si elle est connue.--strain <souche>: spécifier la souche si elle est connue.--gcode <int>: code génétique à utiliser. Par défaut le code standard (0) est utilisé. Cette option va de paire avec le paramètre--kingdom--rfam: activer la recherche des ARN non codants avec Infernal et Rfam (exécution plus longue)--norrna:ne pas annoter les ARNs ribosomiques--notrna: ne pas annoter les ARNs de transfert--compliant: rendre les outputs conformes au format Genbank/ENA/DDJB--centre [identifiant]: spécifier l'identifiant de séquencage (?)--outdir <repertoire>: spécifier le répertoire de sortie.--force: écraser les fichiers dans le répertoire de sortie--cpus <nombre>: nombre de cpus à utiiser à l'éxécution
Pour exécuter Prokka utilisez la forme suivante : prokka [options] --centre X --compliant contigs.fasta
Les outputs de SPAdes se trouvent tous dans le dossier spécifié. La liste non-exhaustive suivante représente quelques uns des fichiers outputs de SPAdes :
- before_rr.fasta – contient les contigs avant résolution du graphe d'assemblage.
- contigs.fasta – contient les contigs
- scaffolds.fasta – contient les scaffolds
- assembly_graph.fastg – contient le graphe d'assemblage de SPAdes en format FASTG
- contigs.paths – contient les chemins présents dans le graphe d'assemblage des contigs
- scaffolds.paths – contient les chemins présents dans le graphe d'assemblage des scaffolds
- params.txt – information sur les paramètres de SPAdes utilisés au cours de l'éxécution
- spades.log – SPAdes log
- dataset.info – fichier de configutation interne
- K<##>/ – dossier contenant les fichiers correspondant à l'éxécution avec comme longueur de kmer <##>Explication de l'entête des fichiers contigs.fasta et scaffolds.fasta
Les entêtes des fichiers fasta en output sont de la forme suivante : >NODE_7_length_217965_cov_40.4652_ID_4150. Le 7 désigne le numéro du noeud au sein du graphe (correspond à un contig ou un scaffold). 217965 désigne la longueur de la séquence, 40.4652 la couverture des kmers et 4150 l'ID unique associé au noeud.
Prokka
Prokka [6] est un pipeline écrit en perl pour l'annotation rapide de génomes prokaryotes. L'annotation des génomes permet l'extraction d'informations génétiques pertinentes de la séquence nucléotidique brute des génomes.
Prokka est installé sur les serveurs du M5. Vous devez tout d'abord charger le module avant toute tentative d'exécution.
module load prokka
La simplicité des commandes de Prokka permet de l'exécuter avec un mimimum de paramètres. Ainsi un simple ``prokka contigs.fa``` suffira pour annoter un génome d'intérêt à partir des contigs contenus dans le fichier contigs.fa. Il est toutefois possible de spécifier soi-même les paramètres, ce qui présente un avantage particulier pour les utilisateurs avancés. Prokka fourni également des modules pour la soumission des données sur Genbank et ENA.
Pour accéder à l'aide, il suffit de faire :prokka --help
prokka [options] <contigs.fasta>Vous trouverez ci-dessous une liste d'options fréquemment utilisées:
--kingdom <Archaea | Bacteria | Mitochondria | Viruses>: permet de spécifier le domaine à utiliser par Prokka parmi ceux proposés. Par défaut, Prokka assume qu'il s'agit d'un génome bactérien.--usegenus: permet d'indiquer s'il faut utiliser ou non une base de données BLAST spécifique à un genre taxonomic. Nécessite l'utilisation du paramètre--genus--genus <genre>: spécifier le genre taxonomic du génome s'il est connu.--species <espèce>: spécifier l'espèce si elle est connue.--strain <souche>: spécifier la souche si elle est connue.--gcode <int>: code génétique à utiliser. Par défaut le code standard (0) est utilisé. Cette option va de paire avec le paramètre--kingdom--rfam: activer la recherche des ARN non codants avec Infernal et Rfam (exécution plus longue)--norrna:ne pas annoter les ARNs ribosomiques--notrna: ne pas annoter les ARNs de transfert--compliant: rendre les outputs conformes au format Genbank/ENA/DDJB--centre [identifiant]: spécifier l'identifiant de séquencage (?)--outdir <repertoire>: spécifier le répertoire de sortie.--force: écraser les fichiers dans le répertoire de sortie--cpus <nombre>: nombre de cpus à utiiser à l'éxécution
Pour exécuter Prokka utilisez la forme suivante : prokka [options] --centre X --compliant contigs.fasta
FastQC
FastQC est un programme JAVA de control de qualité de données de séquençage. FastQC utilise plusieurs métriques pour évaluer la qualité des séquences. Un rapport (html) est produit à la fin de l'analyse. Ce rapport très simple permet d'identifier les types d'erreurs potentiels au sein des données, donnant ainsi une idée sur leur origine. Le programme peut être éxécuté soit en ligne de commande, soit à partir de l'interface graphique.

Pour accéder à l'aide, faites:fastqc --help
fastqc [-o répertoire de sortie] [-f fastq|bam|sam] [-t nombre de threads] [-c fichier de contaminant] sequence1 sequence2 ... sequenceN...